![]()
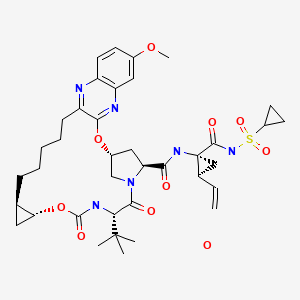
GRAZOPREVIR
- Grazoprevir hydrate
- UNII-4O2AB118LA
- MK 5172
THERAPEUTIC CLAIM Antiviral
CHEMICAL NAMES
1. Cyclopropanecarboxamide, N-[[[(1R,2R)-2-[5-(3-hydroxy-6-methoxy-2-
quinoxalinyl)pentyl]cyclopropyl]oxy]carbonyl]-3-methyl-L-valyl-(4R)-4-hydroxy-L-prolyl-1-
amino-N-(cyclopropylsulfonyl)-2-ethenyl-, cyclic (1→2)-ether, hydrate (1 :1) (1R,2S)-
2. (1aR,5S,8S,10R,22aR)-N-{(1R,2S)-1-[(cyclopropylsulfonyl)carbamoyl]-2-
ethenylcyclopropyl}-5-(1,1-dimethylethyl)-14-methoxy-3,6-dioxo-
1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-
methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-
carboxamide hydrate
MOLECULAR FORMULA C38H50N6O9S.H2O
MOLECULAR WEIGHT 784.92
SPONSOR Merck Sharp & Dohme Corp.
CAS REGISTRY NUMBER 1350462-55-3 HYDRATE, 1350514-68-9 (anhydrous)
WHO NUMBER
9857
GRAZOPREVIR
MERCK
MK-5172 is in phase II clinical development at Merck & Co. for the oral treatment of chronic hepatitis C in combination with peginterferon and ribavirin and in combination with MK-8742. Phase I clinical trials are ongoing for the treatment of hepatitis C in patients with genotype 1 and genotype 3. In 2013, breakthrough therapy designation was assigned to the compound.
Discovery of MK-5172, a macrocyclic hepatitis C virus NS3/4a protease inhibitor
ACS Med Chem Lett 2012, 3(4): 332DOI: 10.1021/ml300017p
Development of a practical, asymmetric synthesis of the hepatitis c virus protease inhibitor MK-5172
Org Lett 2013, 15(16): 4174
References on MK-5172 hydrate:
[1]. Steven Harper , John A. McCauley , Michael T. Discovery of MK-5172, a Macrocyclic Hepatitis C Virus NS3/4a Protease Inhibitor. ACS Med. Chem. Lett., 2012, 3 (4), pp 332-336[2]. Summa V, Ludmerer SW, McCauley JA, MK-5172, a selective inhibitor of hepatitis C virus NS3/4a protease with broad activity across genotypes and resistant variants. Antimicrob Agents Chemother. 2012 Aug;56(8):4161-7.
WO2013142159
WO 2013106631
WO 2013101550
WO 2013028470
WO 2013028471
WO2013028465
WO 2010011566
![]()
Description:
IC50 Value: 7.4nM and 7nM for genotype1b and 1a respectively, in replicon system [1]
MK-5172 is a novel P2-P4 quinoxaline macrocyclic HCV NS3/4a protease inhibitor currently in clinical development.
in vitro: In biochemical assays, MK-5172 was effective against a panel of major genotypes and variants engineered with common resistant mutations observed in clinical studies with other NS3/4a protease inhibitors. In the replicon assay, MK-5172 demonstrated subnanomolar to low-nanomolar EC50s against genotypes 1a, 1b, and 2a [2].
in vivo: In rats, MK-5172 showed a plasma clearance of 28 ml/min/kg and plasma half-life of 1.4 hr. When dosed p.o. at 5 mg/kg, the plasma exposure of MK-5172 was good with an AUC of 0.7 uM.hr. The liver exposure of the compound was quite good (23 uM at 4 hr), and MK-5172 remained in liver 24 hr after a single p.o. 5 mg/kg dose. At 24 hr, the liver concentration of MK-5172 was 0.2 uM, which was over 25-fold higher than the IC50 in the replicon assay with 50% NHS. When dosed to dogs, MK-5172 showed low clearance of 5 ml/min/kg and a 3 hr half-life after i.v. 2 mg/kg dosing and had good plasma exposure (AUC=0.4 uM.hr) after a p.o. 1 mg/kg dose [1].
Clinical trial: Evaluation of Hepatic Pharmacokinetics for MK-5172 in Participants With Chronic Hepatitis C . Phase1
Hepatitis C virus (HCV) infection is a major health problem that leads to chronic liver disease, such as cirrhosis and hepatocellular carcinoma, in a substantial number of infected individuals. Current treatments for HCV infection include immunotherapy with recombinant interferon-α alone or in combination with the nucleoside analog ribavirin.
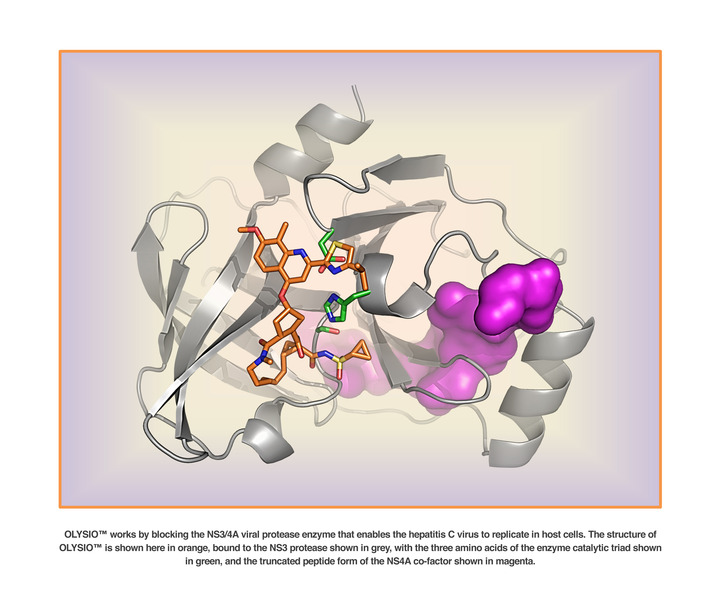
Several virally-encoded enzymes are putative targets for therapeutic intervention, including a metalloprotease (NS2-3), a serine protease (NS3), a helicase (NS3), and an RNA-dependent RNA polymerase (NS5B). The NS3 protease is located in the N-terminal domain of the NS3 protein. NS4A provide a cofactor for NS3 activity.
Potential treatments for HCV infection have been discussed in the different references including Balsano, Mini Rev. Med. Chem. 8(4):307-318, 2008, Rönn et al., Current Topics in Medicinal Chemistry 8:533-562, 2008, Sheldon et al., Expert Opin. Investig. Drugs 16(8):1171-1181, 2007, and De Francesco et al., Antiviral Research 58:1-16, 2003
Different HCV inhibitors are described in different publications. Macrocyclic compounds useful as inhibitors the HCV protease inhibitors are described in WO 06/119061, WO 7/015785, WO 7/016441, WO 07/148,135, WO 08/051,475, WO 08/051,477, WO 08/051,514, WO 08/057,209. Additional HCV NS3 protease inhibitors are disclosed in International Patent Application Publications WO 98/22496, WO 98/46630, WO 99/07733, WO 99/07734, WO 99/38888, WO 99/50230, WO 99/64442, WO 00/09543, WO 00/59929, WO 02/48116, WO 02/48172, British Patent No. GB 2 337 262, and U.S. Pat. No. 6,323,180.
………………………
nmr
![Figure US08080654-20111220-C00021]()
13C NMR (100 MHz, DMSO-d6) δ 172.32, 170.63, 169.04, 159.86, 156.95, 154.74, 148.10, 140.41, 133.55 (2 signals), 128.94, 118.21, 117.58, 105.89, 74.88, 59.75, 58.71, 55.68, 54.13, 54.01, 40.13, 34.49, 34.04, 33.76, 32.68, 30.71, 30.43, 28.55, 27.69, 27.28, 26.38, 21.98, 18.49, 10.67, 5.69, 5.46; MS (ES+) m/z 767 (M+H)+
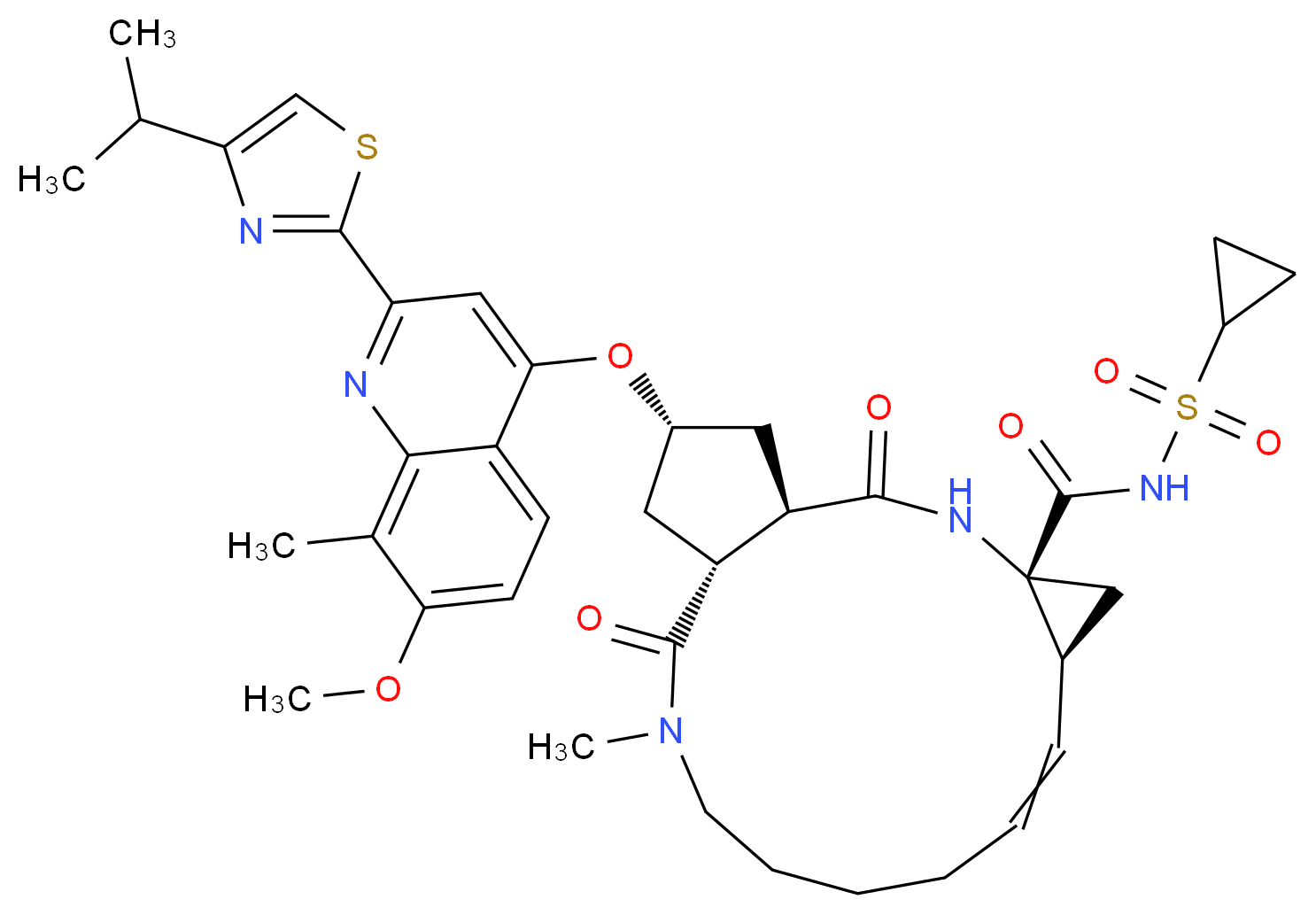
(1aR,5S,8S,10R,22aR)-5-tert-butyl-N-((1R,2S)-1-{[(cyclopropylsulfonyl)amino]carbonyl}-2-vinylcyclopropyl)-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-carboxamide
.…………………
NMR OF GRAZOPREVIR K SALT
Potassium {[(1R,2S)-1-({[(1aR,5S,8S,10R,22aR)-5-tert-butyl-14-methoxy-3,6-dioxo-
1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-
methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxalin-8-
yl]carbonyl}amino)-2-ethenylcyclopropyl]carbonyl}(cyclopropylsulfonyl)azanide (15 K-salt).
1H NMR (400 MHz, DMSO-d6) δ 7.91 (br s, 1 H), 7.75 (d, J =
8.3 Hz, 1 H), 7.15 (m, 1 H), 7.04 (m, 1 H), 5.97 (m, 1 H), 5.73 (br s, 1 H), 4.96 (m, 1 H), 4.79 (apparent q, J = 9.3 Hz, 1 H), 4.26 (dd, J = 9.7, 7.7 Hz, 1 H), 4.20 (d, J = 11.3 Hz, 1 H), 4.14 (d, J = 8.8 Hz, 1 H), 3.90 (dd, J = 11.1, 3.2 Hz, 1 H), 3.86 (s, 3 H), 3.62 (m, 1 H), 2.86-2.60 (m, 3 H), 2.38 (m, 1 H), 2.21 (m, 1 H), 1.80-1.48 (m, 6 H), 1.42 (m, 5 H), 1.14 (m, 1 H), 0.95 (m, 10 H), 0.81 (m, 2 H), 0.72-0.50 (m, 3 H), 0.41 (m, 1 H) ppm.http://pubs.acs.org/doi/suppl/10.1021/ml300017p/suppl_file/ml300017p_si_001.pdf
………………………………………………………
GRAZOPREVIR
(1aR,5S,8S,10R,22aR)-5-tert-Butyl-N-((1R,2S)-1-{[(cyclopropylsulfonyl)amino] carbonyl}-2-
vinylcyclopropyl)-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-
7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-
carboxamide (MK-5172, 15).
1H NMR (400 MHz, CD3
OD) δ 7.79 (dd, J = 9.6, 1.8 Hz, 1 H), 7.23 (s, 1 H), 7.22 (m, 1 H), 7.10 (d, J = 9.6 Hz, 1 H), 6.01 (apparent t, J = 3.6 Hz, 1 H), 5.74 (m, 1 H), 5.24 (dd, J = 17.0 Hz, 1.6 Hz, 1 H), 5.11 (dd, J = 10.4 Hz, 1.6 Hz, 1 H), 4.49 (d, J = 11.2 Hz, 1 H), 4.40 (m, 2 H), 4.13 (dd, J = 12.0 Hz, 4.0 Hz, 1 H), 3.92 (s, 3 H), 3.76 (m, 1 H), 2.92 (m, 2 H), 2.85 (m, 1 H), 2.55 (dd, J = 13.6 Hz, 6.4 Hz, 1 H), 2.28 (m, 1 H), 2.18 (apparent q, J =8.8 Hz, 1 H), 1.85 (dd, J = 8.0 Hz, 5.6 Hz, 1 H), 1.73 (m, 2 H), 1.5 (m, 2 H), 1.40 (dd, J = 9.6 Hz, 5.6 Hz, 1 H), 1.3 (m, 2 H), 1.23 (m, 4 H), 1.08 (s, 9 H), 0.99 (m, 2 H), 0.89 (m, 3 H), 0.73 (m, 1 H), 0.49 (m, 1 H) ppm; HRMS (ESI) m/z 767.3411 [(M+H)+; calcd for C38H51N6O9S: 767.3433].http://pubs.acs.org/doi/suppl/10.1021/ml300017p/suppl_file/ml300017p_si_001.pdf
…………………………..
http://www.google.nl/patents/US8080654
SYNTHESIS OF INTERMEDIATES Intermediates A
|
| Intermediate # |
Structure |
Name |
Lit. Reference |
|
| A1 |
|
(1R,2S)-1-Amino-N- (cyclopropylsulfonyl)-2- vinylcyclopropanecarboxamide hydrochloride |
Wang et al., U.S. Pat. No. 6,995,174 |
|
Intermediate B1 3-methyl-N-({[(1R,2R)-2-pent-4-en-1-ylcyclopropyl]oxy}carbonyl)-L-valine
Step 1: [(1E)-hepta-1,6-dien-1-yloxy](trimethyl)silane
![Figure US08080654-20111220-C00005]()
A solution (0.5 M) of butenyl magnesium bromide in THF (1.4 eq) was treated at −78° C. with Cu(I) Br.SMe2 (0.05 eq) and HMPA (2.4 eq). The mixture was stirred for 10 min, then a solution (1 M) of acrolein (1 eq) and TMSCl (2 eq) in THF was added over 1 h such that the internal temperature remained below −68° C. The resulting mixture was stirred at −78° C. for 2 h, then treated with excess Et3N and diluted with hexane. After reaching room temperature, the mixture was treated with a small portion of H2O and filtered through CELITE. The filtrate was washed 10 times with H2O and then with brine. The organic layer was dried, and the volatiles were removed to give a residue that was distilled under reduced pressure (20 mbar). The fraction collected at 80-86° C. contained the title compound (58%) as a colorless liquid. 1H NMR (400 MHz, CDCl3) δ 6.19 (d, J=11.6 Hz, 1H), 5.85-5.75 (m, 1H), 5.02-4.92 (m, 3H), 2.08-2.02 (m, 2H), 1.94-1.88 (m, 2H), 1.46-1.38 (m, 2H), 0.18 (s, 9H).
Step 2: trans-2-pent-4-en-1-ylcyclopropanol
![Figure US08080654-20111220-C00006]()
A solution (0.45 M) of the preceding compound in hexane was treated with a solution (15%) of Et2Zn (1.2 eq) in toluene, and the resulting solution was cooled in an ice bath. Diiodomethane (1.2 eq) was added dropwise, then the solution was stirred for 1 h before being warmed to 20° C. Pyridine (6 eq) was added, and the slurry was stirred for 15 min then poured onto petroleum ether. The mixture was filtered repeatedly through CELITE until a transparent solution was obtained. This mixture was concentrated at 100 mbar, and the solution that remained (that contained trimethyl{[(trans)-2-pent-4-en-1-ylcyclopropyl]oxy}silane, toluene and pyridine) was further diluted with THF. The mixture was cooled to 0° C. then treated dropwise with a solution (1 M) of TBAF (1.2 eq) in THF. After 10 min, the mixture was allowed to warm to 20° C., and after a further 1 h was poured into H2O. The aqueous phase was extracted with EtOAc, and the combined organic extracts were washed with brine then dried. Removal of the volatiles afforded a residue that was purified by flash chromatography (eluent 0-66% Et2O/petroleum ether) to furnish the title compound (71%) as a colorless liquid. 1H NMR (400 MHz, CDCl3) δ 5.85-5.75 (m, 1H), 5.00 (dd, J=17.1, 1.6 Hz, 1H), 4.94 (br d, J=10.4 Hz, 1H), 3.20 (apparent dt, J=6.4, 2.5 Hz, 1H), 2.10-2.04 (m, 2H), 1.52-1.44 (m, 2H), 1.29-1.19 (m, 1H), 1.15-1.07 (m, 1H), 0.95-0.87 (m, 1H), 0.71-0.66 (m, 1H), 0.31 (apparent q, J=6.0 Hz, 1H).
Step 3: methyl 3-methyl-N-(oxomethylene)-L-valinate
A solution (0.39 M) of methyl 3-methyl-L-valinate in a 2:1 mixture of saturated aqueous NaHCO3 and CH2Cl2 was cooled in an ice bath and stirred rapidly. The mixture was treated with triphosgene (0.45 eq) in one portion, and the resulting mixture was stirred for 0.5 h. The reaction was diluted with CH2Cl2, and the layers were separated. The aqueous phase was extracted with CH2Cl2, then the combined organics were washed with brine and dried. Removal of the solvent gave the title compound as clear oil that was kept for 12 h under vacuum (0.1 mbar) then used directly in the subsequent step. 1H NMR (400 MHz, CDCl3) δ 3.79 (s, 3H), 3.75 (s, 1H), 1.00 (s, 9H).
Step 4: methyl 3-methyl-N-({[(1R,2R)-2-pent-4-en-1-ylcyclopropyl]oxy}carbonyl)-L-valinate and methyl 3-methyl-N-({[(1S,2S)-2-pent-4-en-1-ylcyclopropyl]oxy}carbonyl)-L-valinate
A solution (0.45 M) of trans-2-pent-4-en-1-ylcyclopropanol in toluene was treated with methyl 3-methyl-N-(oxomethylene)-L-valinate (1.1 eq) and then DMAP (1 eq). The resulting mixture was heated under reflux for 12 h then cooled to 20° C. H2O and EtOAc were added, and the organic layer was separated and washed with 1N HCl, brine and dried. Removal of the volatiles afforded a residue that was purified twice by flash chromatography (eluent 0-30% Et2O/petroleum ether). The first fractions contained methyl 3-methyl-N-({[(1R,2R)-2-pent-4-en-1-ylcyclopropyl]oxy}carbonyl)-L-valinate (38%) as an oil. MS (ES+) m/z 298 (M+H)+
The later fractions contained methyl 3-methyl-N-({[(1S,2S)-2-pent-4-en-1-ylcyclopropyl]oxy}carbonyl)-L-valinate (28%) as an oil. MS (ES+) m/z 298 (M+H)+
Step 5: 3-methyl-N-({[(1R,2R)-2-pent-4-en-1-ylcyclopropyl]oxy}carbonyl)-L-valine
A solution (0.1 M) of methyl 3-methyl-N-({[(1R,2R)-2-pent-4-en-1-ylcyclopropyl]oxy}carbonyl)-L-valinate in 2:1 mixture of MeOH/H2O was treated with LiOH.H2O (4 eq) and then heated at 60° C. for 4 h. The mixture was cooled and concentrated to half volume, then diluted with EtOAc and acidified with aqueous HCl (1 N). The organic layer was separated and washed with brine then dried. Removal of the volatiles afforded the title compound (98%) as an oil. MS (ES+) m/z 284 (M+H)+
Intermediates C Intermediate C1 methyl (4R)-4-[(3-chloro-7-methoxyquinoxalin-2-yl)oxy]-L-prolinate hydrochloride
Step 1: 6-methoxyquinoxaline-2,3-diol
A suspension of 4-methoxybenzene-1,2-diamine dihydrochloride in diethyl oxalate (8 eq) was treated with Et3N (2 eq) and then heated at 150° C. for 2 h. The mixture was cooled and filtered, and then the collected solid was washed with H2O and EtOH. The residue was dried to give the title compound (69%). MS (ES+) m/z 193 (M+H)+
Step 2: 3-chloro-6-methoxyquinoxalin-2-ol
A solution (1.53 M) of 6-methoxyquinoxaline-2,3-diol in DMF was treated with SOCl2 (1 eq) and heated at 110° C. After 1.5 h, the reaction mixture was cooled and poured into aqueous HCl (1 N). The resulting precipitate was filtered and washed with H2O and Et2O. The dried solid contained predominantly the title compound as a mixture with 6-methoxyquinoxaline-2,3-diol and 2,3-dichloro-6-methoxyquinoxaline. This material was used directly in the subsequent step. MS (ES+) m/z 211 (M+H)+
Step 3: 1-tert-butyl 2-methyl (2S,4R)-4-[(3-chloro-7-methoxyquinoxalin-2-yl)oxy]pyrrolidine-1,2-dicarboxylate
A solution (0.35 M) of 3-chloro-6-methoxyquinoxalin-2-ol in NMP was treated with Cs2CO3 (1.5 eq) and 1-tert-butyl 2-methyl (2S,4S)-4-{[(4-bromophenyl)sulfonyl]oxy}pyrrolidine-1,2-dicarboxylate (1.1 eq). The resulting mixture was stirred at 50° C. for 18 h, then a further portion (0.1 eq) of 1-tert-butyl 2-methyl (25,45)-4-{[(4-bromophenyl)sulfonyl]oxy}pyrrolidine-1,2-dicarboxylate was added. After stirring for 2 h, the mixture was cooled and diluted with H2O and EtOAc. The organic phases were washed with aqueous HCl (1 N), saturated aqueous NaHCO3 and brine. The dried organic phase was concentrated to a residue that was purified by flash-chromatography (0-60% EtOAc/petroleum ether) to give the title compound (35% for two steps) as a solid. MS (ES+) m/z 438 (M+H)+
Step 4: methyl (4R)-4-[(3-chloro-7-methoxyquinoxalin-2-yl)oxy]-L-prolinate hydrochloride
A solution (0.62 M) of 1-tert-butyl 2-methyl (2S,4R)-4-[(3-chloro-7-methoxyquinoxalin-2-yl)oxy]pyrrolidine-1,2-dicarboxylate in CH2Cl2 was treated with a solution (4 M) of HCl in dioxane (5 eq). The mixture was stirred at 20° C. for 2 h, then treated with a solution (4 M) of HCl in dioxane (2 eq). After 5 h, the reaction was judged complete and the mixture was concentrated under reduced pressure. The residue was triturated with Et2O to give the title compound (95%) as a solid. MS (ES+) m/z 338 (M+H)+
Example 1 Potassium {[(1R,2S)-1-({[(1aR,5S,8S,10R,22aR)-5-tert-butyl-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxalin-8-yl]carbonyl}amino)-2-vinylcyclopropyl]carbonyl}(cyclopropylsulfonyl)azanide
Step 1: methyl 3-methyl-N-({[(1R,2R)-2-pent-4-en-1-ylcyclopropyl]oxy}carbonyl)-L-valyl-(4R)-4-[(3-chloro-7-methoxyquinoxalin-2-yl)oxy]-L-prolinate
A solution (0.2 M) of methyl (4R)-4-[(3-chloro-7-methoxyquinoxalin-2-yl)oxy]-L-prolinate hydrochloride in DMF was treated with 3-methyl-N-({[(1R,2R)-2-pent-4-en-1-ylcyclopropyl]oxy}carbonyl)-L-valine (1.1 eq), DIEA (5 eq) and HATU (1.2 eq). The resulting mixture was stirred at 20° C. for 5 h, then diluted with EtOAc. The organic layer was separated and washed with aqueous HCl (1 N), saturated aqueous NaHCO3 and brine. The dried organic phase was concentrated under reduced pressure to give a residue that was purified by flash chromatography (eluent 10-30% EtOAc/petroleum ether) to furnish the title compound (96%) as an oil. MS (ES+) m/z 604 (M+H)+
Step 2: methyl 3-methyl-N-({[(1R,2R)-2-pent-4-en-1-ylcyclopropyl]oxy}carbonyl)-L-valyl-(4R)-4-[(7-methoxy-3-vinylquinoxalin-2-yl)oxy]-L-prolinate
A solution (0.1 M) of methyl 3-methyl-N-({[(1R,2R)-2-pent-4-en-1-ylcyclopropyl]oxy}carbonyl)-L-valyl-(4R)-4-[3-chloro-7-methoxyquinoxalin-2-yl)oxy]-L-prolinate in EtOH was treated with potassium trifluoro(vinyl)borate (1.5 eq) and triethylamine (1.5 eq). The resulting mixture was degassed, then PdCl2(dppf)-CH2Cl2 adduct (0.1 eq) was added. The mixture was heated under reflux for 1 h, then cooled to room temperature and diluted with H2O and EtOAc. The organic phase was separated, washed with H2O and brine then dried. Removal of the volatiles afforded a residue that was purified by flash chromatography (20-30% EtOAc/petroleum ether) to give the title compound as a yellow foam that was used directly in the subsequent step. MS (ES+) m/z 595 (M+H)+
Step 3: methyl (1aR,5S,8S,10R,18E,22aR)-5-tert-butyl-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,20,21,22,22a-dodecahydro-8H-7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-carboxylate
A solution (0.02 M) of methyl 3-methyl-N-({[(1R,2R)-2-pent-4-en-1-ylcyclopropyl]oxy}carbonyl)-L-valyl-(4R)-4-[(7-methoxy-3-vinylquinoxalin-2-yl)oxy]-L-prolinate in DCE was heated to 80° C. then treated with Zhan 1 catalyst (0.15 eq). The resulting mixture was stirred at 80° C. for 1 h, then cooled to room temperature and concentrated under reduced pressure. The residue was purified by flash chromatography (20-50% EtOAc/petroleum ether) to give the title compound (25% for 2 steps) as a foam. MS (ES+) m/z 567 (M+H)+
Step 4: methyl (1aR,5S,8S,10R,22aR)-5-tert-butyl-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-carboxylate
A solution (0.05 M) of methyl (1aR,5S,8S,10R,18E,22aR)-5-tert-butyl-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,20,21,22,22a-dodecahydro-8H-7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-carboxylate in MeOH/dioxane (1:1 ratio) was treated with Pd/C (8% in weight). The resulting mixture was stirred under atmosphere of hydrogen for 4 h. The catalyst was filtered off, and the filtrate was concentrated under reduced pressure to give the title compound (98%) as a solid. MS (ES+) m/z 569 (M+H)+
Step 5: (1aR,5S,8S,10R,22aR)-5-tert-butyl-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-carboxylic acid
A solution (0.1 M) of methyl (1aR,5S,8S,10R,22aR)-5-tert-butyl-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-carboxylate in a 1:1 mixture of H2O/THF was treated with LiOH.H2O (3 eq). The resulting mixture was stirred at 20° C. for 18 h, acidified with aqueous HCl (0.2 M) and diluted with EtOAc. The organic phase was separated, washed with aqueous HCl (0.2 M) and brine then dried. Removal of the volatiles afforded the title compound (98%) as a solid. MS (ES+) m/z 555 (M+H)+
Step 6: (1aR,5S,8S,10R,22aR)-5-tert-butyl-N-((1R,2S)-1-{[(cyclopropylsulfonyl)amino]carbonyl}-2-vinylcyclopropyl)-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-carboxamide
![Figure US08080654-20111220-C00021]()
A solution (0.1 M) of (1aR,5S,8S,10R,22aR)-5-tert-butyl-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-carboxylic acid in CH2Cl2 was treated with (1R,2S)-1-{[(cyclopropylsulfonyl)amino]carbonyl}-2-vinylcyclopropanaminium chloride (1.3 eq), DIEA (3 eq), DMAP (1.5 eq) and TBTU (1.45 eq). The resulting mixture was stirred at 20° C. for 18 h and then diluted with EtOAc. The solution was washed with aqueous HCl (0.2 M), saturated aqueous NaHCO3 and brine. The organic phases were dried and concentrated to give a residue that was purified by flash-chromatography (eluent 2.5% MeOH/CH2Cl2) to give the title compound (89%) as a solid. 13C NMR (100 MHz, DMSO-d6) δ 172.32, 170.63, 169.04, 159.86, 156.95, 154.74, 148.10, 140.41, 133.55 (2 signals), 128.94, 118.21, 117.58, 105.89, 74.88, 59.75, 58.71, 55.68, 54.13, 54.01, 40.13, 34.49, 34.04, 33.76, 32.68, 30.71, 30.43, 28.55, 27.69, 27.28, 26.38, 21.98, 18.49, 10.67, 5.69, 5.46; MS (ES+) m/z 767 (M+H)+
GRAZOPREVIR POTASSIUM
Step 7: potassium {[(1R,2S)-1-({[(1aR,5S,8S,10R,22aR)-5-tert-butyl-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxalin-8-yl]carbonyl}amino)-2-vinylcyclopropyl]carbonyl}(cyclopropylsulfonyl)azanide
The preceding material was taken up in EtOH and the resulting solution (0.025 M) was cooled to 0° C. A solution (0.02 M) of tert-BuOK (1.5 eq) in EtOH was added leading to the formation of a precipitate. The mixture was stirred at 20° C. for 18 h, then the solid was collected by filtration. This material was washed with EtOH and dried to give the title compound (93%) as a white crystalline solid. MS (ES+) m/z 767 (M+H)+http://www.google.nl/patents/US8080654
Filed under:
phase2 drugs Tagged:
anthony crasto,
GRAZOPREVIR,
medicinal chemistry,
organic chemistry,
phase 2,
world drug tracker ![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()































































































.png)